
 Laurent Schwartz
Laurent Schwartz
[sg_popup id= »6″ event= »hover »]38.09 Assistance Publique – Hôpitaux de Paris[/sg_popup]
 Khalid Omer Alfarouk
Khalid Omer Alfarouk
[sg_popup id= »8″ event= »hover »]20.56 University of California, San Francisco[/sg_popup]
 laudiu T Supuran
laudiu T Supuran
[sg_popup id= »9″ event= »hover »]54.3 University of Florence[/sg_popup]
Table of Contents
Abstract 3
Introduction..4
1) Decrease energy yield of the cancer cells and compensatory fermentation..5
2) Oncogenes and carcinogens increase the Warburg’s effect 6
3) The Warburg’s effect explains intracellular alkalosis and cell proliferation..7
4) Tumor anabolism (the Warburg’s effect) explains the increased pressure and the invasion of surrounding tissue.9
5) Impact of the Warburg’s effect and subsequent increased pressure on the surrounding normal cells and immune response.10
6) Human cancer growth is nonlinear, because of progressively decreasing oxidative phosphorylation 12
7) Metabolic rewiring: the combination of alpha Lipoic acid and hydroxycitrate slows cancer growth, whatever the primary site.13
Conclusion: 15
Abstract
It is a longstanding debate whether cancer is one disease or a set of very diverse diseases. The goal of this paper is to suggest strongly that most (if not all) the hallmarks of cancer could be the consequence of the Warburg‘s effect. As a result of the metabolic impairment of the oxidative phosphorylation, there is a decrease in ATP concentration. To compensate the reduced energy yield, there is massive glucose uptake, anaerobic glycolysis, with an up-regulation of the Pentose Phosphate Pathway resulting in increased biosynthesis leading to increased cell division and local pressure. This increased pressure is responsible for the fractal shape of the tumor, the secretion of collagen by the fibroblasts and plays a critical role in metastatic spread. The massive extrusion of lactic acid contributes to the extracellular acidity and the activation of the immune system. The decreased oxidative phosphorylation leads to impairment in CO2 levels inside and outside the cell, with increased intracellular alkalosis and contribution of carbonic acid to extracellular acidosis-mediated by at least two cancer-associated carbonic anhydrase isoforms. The increased intracellular alkalosis is a strong mitogenic signal, which bypasses most inhibitory signals. Mitochondrial disappearance (such as seen in very aggressive tumors or following chemotherapy) is a consequence of impaired mitochondrial yield. Mitochondrial swelling, itself a result of decreased ATP concentration.
The hallmarks of cancer are the consequence of the Warburg effect. Accordingly treatment should aim at restoring the oxidative phosphorylation
Keywords: Warburg’s effect, unification theory, ATP, metabolic treatment, alpha lipoïc acid.
Introduction
Cancer is widely considered as a large variety of diseases with different prognoses, sites of origin, patterns of spread, and kinetics. The goal of this paper is to address the possible underlying unity of this very diverse disease. There is always arguing about the very nature of cancer. “The Hallmarks of Cancer” [1] is a seminal article published in Cell by Hanahan and Weinberg.The authors believe that the complexity of cancer can be reduced to a small number of underlying principles. The paper argues that all cancers share six common traits (“hallmarks”) that govern the transformation of normal cells to cancer cells (malignant or tumor) [2]. These hallmarks stipulate that cancer cells [2]:
- Stimulate their own growth.
- Resist inhibitory signals that might otherwise stop their growth.
- Resist their own programmed cell death.
- Stimulate the growth of blood vessels to supply nutrients to tumors [3].
- Can multiply forever [3].
- Invade local tissue and spread to distant sites
In an update published in 2011, Hanahan and Weinberg [2] proposed two new hallmarks: - Abnormal metabolic pathways.
- Evading the immune system.
The goal of this paper is to suggest strongly that these eight features, as diverse as they may appear, may have a common cause, a decreased oxidative phosphorylation [4].
In the early 1920’s Otto Warburg had demonstrated a unique feature of cancer cells, namely an increased uptake of glucose and secretion of lactic acid by cancer cells, even in the presence of oxygen. This aerobic fermentation is the signature of cancer. Warburg also noticed a concomitant decreased number of mitochondria (grana) [4].The goal of this paper is to link the Warburg‘s effect with some of the hallmarks of cancer (See figure 1).
1) Decrease energy yield of the cancer cells and compensatory fermentation
pite increased glucose uptake, there is a 50% drop in ATP level in human colon cancer cells compared to adjacent benign cells [5]. This decrease in ATP is a consequence of impairment of the oxidative phosphorylation [4,6]. In normal, differentiated cells, the yield of a molecule of glucose is 34 ATP. In cells with functional mitochondria, this energy is derived mostly from oxidative phosphorylation were approximately 88% of total cellular energy is produced [7,8].The other approximate 12% of energy is produced about equally from substrate level phosphorylation through glycolysis in the cytoplasm and the TCA cycle in the mitochondrial matrix [7,8].
This yield drops to 2 ATP when the mitochondrion is turned off (anaerobic glycolysis).To compensate for the decreased energy yield, the cell increases the glucose uptake [7,9]. A consequence of the decreased activity of the mitochondrion is an increased secretion of lactic acid and an activation of the pentose phosphate pathway (PPP).
The activation of the Pentose Phosphate Pathway results from an increase in glucose uptake with a concomitant obstacle downstream of the pentose phosphate shunt, most probably at the level of the pyruvate dehydrogenase[7]and/or of the pyruvate kinase[10]. The increased flux in the pentose phosphate pathway results in:
- A shift toward anabolism due to increased synthesis of NADPH that plays a crucial role in NDPH/NADP+ ratio that determines the redox state of the cell via removal of reactive oxygen species (ROS) and so prevents the cellular death, and so it controls the cellular fate[11–13].
- The shift toward the pentose pathway also results in the production of ribose-5-phosphate, required for the synthesis of nucleic acids [14].
2) Oncogenes and carcinogens increase the Warburg’s effect
When Rous discovered that a virus could transmit cancer, one may have thought that cancer was a viral disease [4,9]. It appeared later that the virus had only transmitted a gene (Src), encoding a non-receptor tyrosine kinase, captured by the virus from a previous host. When it was found that the captured and then retransmitted gene could cause cancer, like many other cellular genes (proto- oncogenes), only because they were up-regulated, one may have thought that cancer was a genetic disease, linked to the oncogenic cellular concept.
However, the Src gene; like all the later discovered oncogenes encode proteins associated with signaling routes, regulating specific metabolic pathways[7]. Since then, some thirty different oncogenes all target and stimulate the anabolic pathways [7].
However, as Warburg wrote in 1956 [4,15], “The chicken Rous sarcoma, which is labeled today as a virus tumor, ferments glucose, and lives as a partial anaerobe like all tumors.” In 2000, Reshkin et al. did transfect the Human Papilloma Virus (HPV) to normal cells, and they found that those cells did overexpress the NHE1 protein which was accompanied by intracellular alkalinity which was first even of malignant transformation [16] while intracellular alkalinity does invest on Warburg Effect [17]
Carcinogenesis, whether arising from viral infection, oncogene activation or chemical agent, produces similar impairment in respiratory enzyme activity and mitochondrial function [8,18,19]. Thus, viruses can potentially cause cancer through displacement of respiration with substrate level phosphorylation in the infected cells [7,8,18,20].
Infection by an oncogenic virus or exposure to a carcinogen inhibits the mitochondria and causes the Warburg’s effect[21–25]. This Warburg’s effect is responsible for the activation of the Pentose phosphate pathway and subsequent anabolism [6,9,20].
As stated by Seyfried [20],” any unspecific condition that damages a cell’s respiratory capacity but is not severe enough to kill the cell can potentially initiate the path to cancer [26]. Some of the many unspecific conditions that can diminish a cell’s respiratory capacity thus initiating carcinogenesis include inflammation, carcinogens, radiation, intermittent hypoxia, rare germline mutations, viral infections, and age”[26].
3) The Warburg’s effect explains intracellular alkalosis and cell proliferation
The inhibition of the oxidative phosphorylation results in the activation of the anabolic pathway such as the pentose phosphate pathway that is necessary for DNA and RNA synthesis [27]. It reduces NADP+ into NADPH required for lipid synthesis and membrane elongation. The decreased mitochondrial activity has a second consequence: cytoplasm alkalinization because of decreased CO2 secretion [28].
Deregulated pH is emerging as another hallmark of cancer because tumors show a ‘reversed’ pH gradient with a constitutively increased intracellular pH that is higher than the extracellular pH. This gradient enables cancer progression by promoting proliferation, the evasion of apoptosis, metabolic adaptation, migration, and invasion [29–32].
Most research has focused on the extracellular acidosis. It is caused by massive lactic acid extrusion and activity of at least two carbonic anhydrases which hydrate CO2 to bicarbonate (shuttled back to the cytosol through NBCs, AEs, etc.) and protons (which remain in the extracellular space) [33]. There is evidence that an acidic extracellular pH promotes invasiveness and metastatic behavior in several tumor models [34–36], proteolytic enzyme activation and matrix destruction [37,38]. Interference with pH regulation in tumors was proposed as an anticancer strategy with at least a Mab and one small molecule sulfonamide (SLC-0111) in clinical trials for the treatment of metastatic solid tumors [39,40].
In normal cells, the intracellular pH oscillates during the cell cycle between 6.8 and 7.3 [41]. The pH is maximum (7.3) before mitosis and decreases to a minimum at S phase (6.8). The oscillation of the pH during the cell cycle matches the value of the decompaction of the histones, the RNA polymerase activation, the DNA polymerase activation and the DNA compaction before mitosis. The main reason for this oscillation is ATP hydrolysis [42] and the subsequent release of proton ions. This oscillation is buffered by several mechanisms such as the Na+/H+ exchanger (NHE-1), the carbonic anhydrases, the proton-linked monocarboxylate transporter, the Cl–/HCO3– exchangers [35,36,42–50]. The increased activity of these transporters buffers the change of pH caused mostly by ATP synthesis and hydrolysis [36].
The intracellular pH of the cancer cells has been less studied. During the cell cycle, it oscillates between 7.2 and 7.5. Intracellular alkalosis is probably a consequence of the decreased oxidative phosphorylation and a decreased secretion of CO2. Carbon dioxide reacts with water to create carbonic acid.Cell transformation or enhanced cancer cell division and resistance to chemotherapy are associated with a more alkaline pH [47–51].
Several effective cancer treatments decrease the intracellular pH. There is extensive literature on increased survival support for the combined use of antacids (which prevent the extrusion proton from the tumor cells) with standard chemotherapy [46,49,52]. Hyperthermia [53] decreases the intracellular pH. Chemotherapy and radiation therapy result in decreased ATP/ADP ratio and intracellular acidification [49,52].
The calorie restricted ketogenic diet [26] will reduce the availability of glucose, the principal metabolite for glycolysis and the PPP. It results in increased level of acidic ketone bodies that cannot be metabolized by the cancer cells (to the difference of normal cells) and probably decrease the intracellular pH. Similarly, acid diet, in general, exogenous lactate, regresses tumor growth [54]. Exogenous lactate leads to spontaneous regression of cancer [55], one of the possible explanation is that lactate might prevent further efflux of the intracellularly produced lactate; therefore lactate will accumulate intracellularly that push the reversible reaction towards formation of TCA. In other words, based on the reversible reaction kinetics, the exogenous lactate might force the activation of mitochondrial respiration.
4) Warburg’s effect is consistent with explaining the increasing pressure and the invasion of surrounding tissue
The word “cancer” comes from the Greek word carcinos (crab). Unlike benign tumors, cancer is harder and irregular. It has a stellar, fractal shape and Cancer invades the surrounding tissue.
In the confined environment of an organ, anabolic growth of cancer cells results in increased pressure [9,56–59]. This increased pressure results in the invasion of surrounding tissues, destruction of blood vessels and distant metastasis [9,56–59].
During a liver biopsy, Coldwell [56] measured the interstitial pressure of the normal hepatic parenchyma, as well as cirrhosis and liver cancer with a needle, advanced to the tumor under CT guidance. The pressure was then obtained when the needle entered the periphery of the tumor. The pressure measured by biopsy demonstrates that the pressure in the normal liver parenchyma is 4 mm Hg. The pressure increases to between 16 and 25 in liver tumors (either primary or metastatic). This increased pressure in the tumor explains the peculiar drainage. The tumors are fed by the hepatic artery and not from the portal vein as the rest of the liver parenchyma. The intra-arterial pressure being 137 mm Hg, it is markedly higher than the mean portal pressure: 4 mm Hg. Only the arterial flow and not the portal flow can reach the liver tumor [56].
Normal cells and especially epithelial cells are organized along a structural axis [57], which allows for cell adhesion to the mesenchyme on one side and the epithelium function on the lumen side [60]. Cancer cells have decreased levels of cadherins and annexins resulting in the loss of cell polarity. They can change plane and thus, escape the physical constraints [57]. Cancer cells can diffuse in areas of lesser physical constraints and invade soft tissue. Cancer cells, unlike normal cells, escape the physical constraints [58,59].
The stellar shape of cancer, such as seen on mammography, can only be explained by increased pressure and the loss of cell polarity [57].
5) Impact of the Warburg’s effect and subsequent increased pressure on the surrounding normal cells and immune response
Inflammation is characterized by tumor, dolor, rubor, and color, as stated by Galen two thousand years ago. Inflammation can be caused by factors as diverse as heat, freezing temperature, trauma or various chemicals [61,62].
Inflammation is a key risk factor for cancer [63]. For example, a virus, alcoholic liver disease, antitrypsin deficiency, hemochromatosis, and tyrosinemia [64] or a direct trauma all can cause hepatitis. Cirrhosis is the complication of untreated hepatitis. Liver cancer is frequently associated with such pre-existing inflammation and fibrosis. Between 60% and 90% of hepatocellular carcinoma occurs in patients with hepatic macronodular cirrhosis[7,56,63].
When a foreign body, such as a splinter, is inserted into the epidermis, there is no inflammation. When this splinter reaches the dermis where the capillaries lay, there is inflammation. Vascular leakage is a common feature of inflammation. It can be caused by direct damage, resulting from a foreign body, burn or necrosis. Whatever the reason for the leakage may be, this results in the leakage of red blood cells, leukocytes, and plasmatic proteins. These proteins will, in turn, be broken down into smaller pieces by the metabolic enzymes. This partial digestion will release a larger amount of osmoles, further increasing the extracellular osmolarity [61,62,65].
Similarly, a high concentration of protein occurs in ascites during inflammation, such as tuberculosis or pancreatitis [7,61,65–71]. The same goes for pericarditis [67,68], atherosclerosis, arthritis or asthma and pneumonia [66–71]. Whenever inflammation occurs, there appears to be an increase of protein in the extracellular space.
The increased pressure induces the secretion of proteases, growth factors, and growth factor receptors. It results in oncogene activation and shifts the cellular metabolism toward anabolism [61,65,72,73].
Anabolism occurs in cancer, just like in inflammation. But, unlike inflammation, cancer also involves neoangiogenesis[7]. Cancer development leads to the formation of an extensive network of new blood vessels. The growth is promoted by growth factors such as VEGF, which in turn is a possible consequence of increased osmolarity[74]. Unlike normal blood angiogenesis during embryogenesis[75], these blood vessels are leaky and hence increase the osmotic pressure as well as the harshness of the tumor. Also, these vessels are synthesized by incorporation of normal endothelial cell with malignant cells « tumor blood vessels mosaicism » [76,77] The increased pressure has an impact on the surrounding normal cells (stroma). It is responsible for extracellular matrix deposition and fibroblast proliferation [74,75,78].
Increased pressure controls the NF-KB activity through the methylation of PP2A [61,67]. More specifically, NF-KB controls the transcription of cytokines and genes that regulate cellular differentiation, survival, and proliferation, thereby controlling various aspects of innate and adaptive immune responses [79]. Increased pressure is responsible for chemokine-cytokine secretion thus contributing to the immune response of cancer [79]. The half-life of monocytes exposed to hyperosmolarity (as seen in the tumor interstitial fluid) doubled the half-life of monocytes and macrophages [61,67].
The secretion of lactic acid results in an extracellular acidicpH.Investigations on polymorphonuclear leukocytes demonstrate mainly inhibition of chemotaxis, respiratory activity, and bactericidal capacity at reduced pH. Evidence of impaired lymphocyte cytotoxicity and proliferation at acidic pH is also beginning to emerge [80–82].
6) Progressive decreasing oxidative phosphorylation might explain why cancer growth is nonlinear
Cancer often arises from the transformation of a benign disease such as polyps. It can also start as a low-grade disease such as chronic myeloid leukemia or a low-grade glioma to a more aggressive acute leukemia or a glioblastoma. These more aggressive cancers have a higher glucose uptake such as seen on PET scan[7,83–85].It is probable that this change in pattern is linked to a decreased mitochondrial activity.
Cytotoxic chemotherapy also influences human cancer growth. Cytotoxic drugs have been selected “in vivo” to kill cancer cells. The antineoplastic drugs are capable of generating a variety of free radical species in subcellular systems, and this capacity has been considered critical for its antitumor action [86–88].
Chemotherapy has had tremendous benefits for pediatric or Hodgkin’s patients. However, for most solid tumors, there is a sizable response rate (complete and partial regression) but limited gain in survival.
At the time of failure of chemotherapy, there is a sharply increased glucose uptake such as seen on PET scan [83]. Resistance to chemotherapy has been correlated with decreased oxidative phosphorylation[7,87–89]and alkaline pH [55].
Cytotoxic drugs injure or even destroy the mitochondria [90,91]. The oxidative phosphorylation is further reduced; the pH is more alkaline, and the PPP is activated. Cancer grows unrelentlessly, and further chemotherapy is usually ineffective[55].
Elliott analyzed, using an electron microscope, the presence of mitochondria in 346 human breast cancer samples. The mitochondria were present in low-grade cancer but spare or absent in the most aggressive tumors [85].
It is probable that this change of behavior from slow-growing, low-grade to high-grade cancer or from cancer that is inhibited by chemotherapy to resistant to cytotoxic drugs is the consequence of increased oxidative phosphorylation.
Mitochondria are fragile. Decrease in ATP result in further mitochondrial damages. The decrease concentration of ATP (4) may result in further damage and swelling of the mitochondria [91,92]. The mitochondria increase in size and the cristae disappear[93–95].
7) Metabolic rewiring: the combination of alpha- lipoïc acid and hydroxycitrate slows cancer growth, whatever the primary site
The decrease in concentration of ATP can be the consequence of decreased availability of acetyl-CoA to the mitochondria and the subsequent rewiring of the metabolic fluxes upstream of the mitochondria [6,9].
Michelakis [96] reported that dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase kinase, resulted in the activation of the mitochondria [64], in vitro and tumor regression in three out of five patients with glioblastoma.
The most likely mechanism of action for α-lipoic acid for its inhibition of tumor growth is the inhibition of pyruvate dehydrogenase kinase (the same target that DCA). This enzyme inhibits the activity of pyruvate dehydrogenase and is known to be up-regulated in cancer cells expressing the aerobic glycolytic phenotype. Pyruvate dehydrogenase (PDH) catalyzes the conversion of pyruvate to acetyl-CoA, the initial step of the ultimate conversion of glucose to carbon dioxide and water, with the concomitant production of ATP, in the TCA cycle. Therefore, it is reasonable to suggest that blocking the activity of pyruvate dehydrogenase kinase will at least partially restore the activity of pyruvate dehydrogenase, thereby increasing the flux of pyruvate through the TCA cycle in the mitochondria, while simultaneously reducing the production of lactic acid and most importantly decreasing the flux in the pentose pathway shunt[64].
A combination of alpha lipoïc acid and hydroxycitrate (an inhibitor of the citrate lyase) [64,97–99] has been reported to slow cancer growth, in murine xenografts. This inhibition appears to be universal (i.e. independent of the primary site) and has been reproduced in different laboratories. Similar data have been obtained with CPI 613 a novel alpha lipoïc acid analog [100]
The first patient diagnosed with a large peritoneal metastasis of a colon carcinoma survived five years after a treatment combining chemotherapy with hydroxycitrate and alpha lipoïc acid [101,102].
Starting January 2013, compassionate metabolic treatment (alpha lipoic acid hydroxycitrate and low dose Naltrexone) was offered to patients sent home after the failure of conventional cytotoxic chemotherapy for metastatic cancer (whatever the primary site) but with a Karnovsky status above 70. Of the first unselected eleven patients, five are alive and reasonably well 30 months after the start of treatment [102]. The second group of patients, having failed first and second line chemotherapy were treated with metabolic treatment and conventional chemotherapy. Survival at one year was 70% [102–104].
These results are in line with recent molecular biology experiments. The introduction of normal mitochondria into cancer cells restore mitochondrial function and inhibit cancer cell growth, and reverse chemoresistance [105–107]. The fusion of cancer cells with normal mitochondria results in increased ATP synthesis, oxygen consumption, and respiratory chain activities [63], marked decrease in cancer growth, resistance to the anti-cancer drug, invasion, and colony formation in soft agar, and « in vivo » tumor growth in nude mice [5,106].
Conclusion:
Today, cancer is thought to be a set of very complex diseases with thousands of different mutations. That apparent complexity has led to personalized medicine. The fact that the combination of alpha lipoïc acid and hydroxycitrate slows down cancer growth in every tumor model suggests that at least some targets are the same in a large spectrum of tumors.
“Cancer, above all other diseases, has countless secondary causes. But, even for cancer, there is only one prime cause. Summarized in a few words, the prime cause of cancer is the replacement of the respiration of oxygen in normal body cells by a fermentation of sugar. All normal body cells meet their energy needs by respiration of oxygen, whereas cancer cells meet their energy needs in great part by fermentation. All normal body cells are thus obligate aerobes, whereas all cancer cells are partial anaerobes [18,108].”
To decrease anabolism, glucose uptake should be reduced, and the oxidative phosphorylation should be restored.
Declarations
Authors’ Contribution:
LS conceived the study and drafted and LS KO and CS designed the manuscript. All authors read and approved the final manuscript.
Conflict of Interest statement:
The authors declare that they have no competing interests.
References:
- Hanahan, D.; Weinberg, R. A. The hallmarks of cancer. Cell 2000, 100, 57–70.
- Hanahan, D.; Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–74.
- Wang, E.; Zaman, N.; Mcgee, S.; Milanese, J.-S.; Masoudi-Nejad, A.; O’Connor-McCourt, M. Predictive genomics: a cancer hallmark network framework for predicting tumor clinical phenotypes using genome sequencing data. Semin. Cancer Biol. 2015, 30, 4–12.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–14.
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R. G.; Mercier, I.; Martinez-Outschoorn, U. E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M. P. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–84.
- Vander Heiden, M. G.; Cantley, L. C.; Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033.
- Maurice Israel, L. S. Cancer: a dysmethylation syndrome?; John Libbey Eurotext, 2005.
- Seyfried, T. N.; Shelton, L. M. Cancer as a metabolic disease. Nutr. Metab. (Lond). 2010, 7, 7.
- Israël, M.; Schwartz, L. The metabolic advantage of tumor cells. Mol. Cancer 2011, 10, 70.
- Mazurek, S.; Boschek, C. B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–8.
- Neumann, C.; Boubakari; Grünert, R.; Bednarski, P. J. Nicotinamide adenine dinucleotide phosphate-regenerating system coupled to a glutathione-reductase microtiter method for determination of total glutathione concentrations in adherent growing cancer cell lines. Anal. Biochem. 2003, 320, 170–8.
- Ganea, E.; Harding, J. J. Glutathione-related enzymes and the eye. Curr. Eye Res. 2006, 31, 1–11.
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206.
- Alfarouk, K. O. Tumor metabolism, cancer cell transporters, and microenvironmental resistance. J. Enzyme Inhib. Med. Chem. 2016, 1–8.
- Warburg, O. On the formation of lactic acid with growth. Biochem. Z. 1925, 160, 307–311.
- Reshkin, S. J.; Bellizzi, A.; Caldeira, S.; Albarani, V.; Malanchi, I.; Poignee, M.; Alunni-Fabbroni, M.; Casavola, V.; Tommasino, M. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 2000, 14, 2185–97.
- Alfarouk, K. O.; Verduzco, D.; Rauch, C.; Muddathir, A. K.; Adil, H. H. B.; Elhassan, G. O.; Ibrahim, M. E.; David Polo Orozco, J.; Cardone, R. A.; Reshkin, S. J.; Harguindey, S. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777–802.
- Roskelley, R. C.; Mayer, N.; Horwitt, B. N.; Salter, W. T. Studies in cancer. vii. Enzyme deficiency in human and experimental cancer. J. Clin. Invest. 1943, 22, 743–51.
- López-Lázaro, M. The warburg effect: why and how do cancer cells activate glycolysis in the presence of oxygen? Anticancer. Agents Med. Chem. 2008, 8, 305–12.
- Seyfried, T. Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; 1st ed.; 2012.
- D’Agostino, D. M.; Bernardi, P.; Chieco-Bianchi, L.; Ciminale, V. Mitochondria as functional targets of proteins coded by human tumor viruses. Adv. Cancer Res. 2005, 94, 87–142.
- Parkin, D. M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–44.
- Clippinger, A. J.; Bouchard, M. J. Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J. Virol. 2008, 82, 6798–811.
- Koike, K. Hepatitis B virus X gene is implicated in liver carcinogenesis. Cancer Lett. 2009, 286, 60–8.
- Pacini, N.; Borziani, F. Cancer stem cell theory and the warburg effect, two sides of the same coin? Int. J. Mol. Sci. 2014, 15, 8893–930.
- Seyfried, T. N.; Flores, R. E.; Poff, A. M.; D’Agostino, D. P. Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis 2014, 35, 515–27.
- Ortega, A. D.; Sánchez-Aragó, M.; Giner-Sánchez, D.; Sánchez-Cenizo, L.; Willers, I.; Cuezva, J. M. Glucose avidity of carcinomas. Cancer Lett. 2009, 276, 125–35.
- da Veiga Moreira, J.; Peres, S.; Steyaert, J.-M.; Bigan, E.; Paulevé, L.; Nogueira, M. L.; Schwartz, L. Cell cycle progression is regulated by intertwined redox oscillators. Theor. Biol. Med. Model. 2015, 12, 10.
- Alfarouk, K. O.; Muddathir, A. K.; Shayoub, M. E. A. Tumor acidity as evolutionary spite. Cancers (Basel). 2011, 3, 408–14.
- Gatenby, R. A.; Gillies, R. J. A microenvironmental model of carcinogenesis. Nat. Rev. Cancer 2008, 8, 56–61.
- Webb, B. A.; Chimenti, M.; Jacobson, M. P.; Barber, D. L. Dysregulated pH: a perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677.
- Lee, C. H.; Cragoe, E. J.; Edwards, A. M. Control of hepatocyte DNA synthesis by intracellular pH and its role in the action of tumor promoters. J. Cell. Physiol. 2003, 195, 61–9.
- Neri, D.; Supuran, C. T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–77.
- Rofstad, E. K. Acidic Extracellular pH Promotes Experimental Metastasis of Human Melanoma Cells in Athymic Nude Mice. Cancer Res. 2006, 66, 6699–6707.
- Cardone, R. A.; Casavola, V.; Reshkin, S. J. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 2005, 5, 786–95.
- Huber, V.; De Milito, A.; Harguindey, S.; Reshkin, S. J.; Wahl, M. L.; Rauch, C.; Chiesi, A.; Pouysségur, J.; Gatenby, R. A.; Rivoltini, L.; Fais, S. Proton dynamics in cancer. J. Transl. Med. 2010, 8, 57.
- Kato, Y.; Lambert, C. A.; Colige, A. C.; Mineur, P.; Noël, A.; Frankenne, F.; Foidart, J.-M.; Baba, M.; Hata, R.-I.; Miyazaki, K.; Tsukuda, M. Acidic extracellular pH induces matrix metalloproteinase-9 expression in mouse metastatic melanoma cells through the phospholipase D-mitogen-activated protein kinase signaling. J. Biol. Chem. 2005, 280, 10938–44.
- Montcourrier, P.; Mangeat, P. H.; Salazar, G.; Morisset, M.; Sahuquet, A.; Rochefort, H. Cathepsin D in Breast Cancer Cells Can Digest Extracellular Matrix in Large Acidic Vesicles. Cancer Res. 1990, 50, 6045–6054.
- Lou, Y.; McDonald, P. C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahmadi, A.; Kyle, A.; Auf dem Keller, U.; Leung, S.; Huntsman, D.; Clarke, B.; Sutherland, B. W.; Waterhouse, D.; Bally, M.; Roskelley, C.; Overall, C. M.; Minchinton, A.; Pacchiano, F.; Carta, F.; Scozzafava, A.; Touisni, N.; Winum, J.-Y.; Supuran, C. T.; Dedhar, S. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011, 71, 3364–76.
- Supuran, C. T. How many carbonic anhydrase inhibition mechanisms exist? J. Enzyme Inhib. Med. Chem. 2016, 31, 345–60.
- Karagiannis, J.; Young, P. G. Intracellular pH homeostasis during cell-cycle progression and growth state transition in Schizosaccharomyces pombe. J. Cell Sci. 2001, 114, 2929–2941.
- Racker, E. Bioenergetics and the problem of tumor growth. Am. Sci. 60, 56–63.
- Lu, X.; Qin, W.; Li, J.; Tan, N.; Pan, D.; Zhang, H.; Xie, L.; Yao, G.; Shu, H.; Yao, M.; Wan, D.; Gu, J.; Yang, S. The growth and metastasis of human hepatocellular carcinoma xenografts are inhibited by small interfering RNA targeting to the subunit ATP6L of proton pump. Cancer Res. 2005, 65, 6843–9.
- Spugnini, E. P.; Citro, G.; Fais, S. Proton pump inhibitors as anti vacuolar-ATPases drugs: a novel anticancer strategy. J. Exp. Clin. Cancer Res. 2010, 29, 44.
- Supuran, C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–81.
- Fais, S.; De Milito, A.; You, H.; Qin, W. Targeting vacuolar H+-ATPases as a new strategy against cancer. Cancer Res. 2007, 67, 10627–30.
- Gillies, R. J.; Martinez-Zaguilan, R.; Martinez, G. M.; Serrano, R.; Perona, R. Tumorigenic 3T3 cells maintain an alkaline intracellular pH under physiological conditions. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 7414–8.
- Moolenaar, W. H. Effects of growth factors on intracellular pH regulation. Annu. Rev. Physiol. 1986, 48, 363–76.
- Simon, S.; Roy, D.; Schindler, M. Intracellular pH and the control of multidrug resistance. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 1128–32.
- Pouysségur, J.; Dayan, F.; Mazure, N. M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443.
- Alfarouk, K. O.; Stock, C.-M.; Taylor, S.; Walsh, M.; Muddathir, A. K.; Verduzco, D.; Bashir, A. H. H.; Mohammed, O. Y.; Elhassan, G. O.; Harguindey, S.; Reshkin, S. J.; Ibrahim, M. E.; Rauch, C. Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 71.
- Cosentini, E.; Haberl, I.; Pertschy, P.; Teleky, B.; Mallinger, R.; Baumgartner, G.; Wenzl, E.; Hamilton, G. The differentiation inducers phenylacetate and phenylbutyrate modulate camptothecin sensitivity in colon carcinoma cells in vitro by intracellular acidification. Int. J. Oncol. 2001, 19, 1069–74.
- Hofer, K. G.; Mivechi, N. F. Tumor cell sensitivity to hyperthermia as a function of extracellular and intracellular pH. J. Natl. Cancer Inst. 1980, 65, 621–5.
- Harguindey, S.; Henderson, E. S.; Naeher, C. Effects of systemic acidification of mice with Sarcoma 180. Cancer Res. 1979, 39, 4364–4371.
- Harguindey, S.; Orive, G.; Luis Pedraz, J.; Paradiso, A.; Reshkin, S. J. The role of pH dynamics and the Na+/H+ antiporter in the etiopathogenesis and treatment of cancer. Two faces of the same coin–one single nature. Biochim. Biophys. Acta 2005, 1756, 1–24.
- Schwartz, L.; Coldwell, D. Is Liver Disease Caused by Increased Pressure? Interstitial Pressure as a Causative Mechanism in Carcinogenesis and in the Differential Blood Supply in Liver Tumors from the Hepatic Artery. J. Liver 2014, 3.
- Fleury, V.; Schwartz, L. Numerical investigation of the effect of loss of cellular polarity on cancer invasiveness and geometry. Fractals 2003, 11, 397–414.
- Baish, J. W.; Jain, R. K. Fractals and cancer. Cancer Res. 2000, 60, 3683–8.
- Tan, D. S.; Potts, H. W.; Leong, A. C.; Gillett, C. E.; Skilton, D.; Harris, W. H.; Liebmann, R. D.; Hanby, A. M. The biological and prognostic significance of cell polarity and E-cadherin in grade I infiltrating ductal carcinoma of the breast. J. Pathol. 1999, 189, 20–7.
- Schwartz, L. Cancer — Between Glycolysis and Physical Constraint. In Cancer — Between Glycolysis and Physical Constraint; Springer Berlin Heidelberg: Berlin, Heidelberg, 2004; pp. 107–116.
- Schwartz, L.; Guais, A.; Pooya, M.; Abolhassani, M. Is inflammation a consequence of extracellular hyperosmolarity? J. Inflamm. (Lond). 2009, 6, 21.
- CASSELTON, P. J.; STACEY, J. L. OBSERVATIONS ON THE NITROGEN METABOLISM OF PROTOTHECA KRUGER. New Phytol. 1969, 68, 731–749.
- Levy Nogueira, M.; da Veiga Moreira, J.; Baronzio, G. F.; Dubois, B.; Steyaert, J.-M.; Schwartz, L. Mechanical Stress as the Common Denominator between Chronic Inflammation, Cancer, and Alzheimer’s Disease. Front. Oncol. 2015, 5, 197.
- Schwartz, L., M. Israël, I. P. Inflammation and Carcinogenesis: A Change in the Metabolic Process. In Cancer Microenvironment and Therapeutic Implications; Baronzio, G.; Fiorentini, G.; Cogle, C. R., Eds.; Springer Netherlands: Dordrecht, 2009; pp. 3–18.
- Abolhassani, M.; Wertz, X.; Pooya, M.; Chaumet-Riffaud, P.; Guais, A.; Schwartz, L. Hyperosmolarity causes inflammation through the methylation of protein phosphatase 2A. Inflamm. Res. 2008, 57, 419–29.
- Runyon, B. A. Malignancy-related ascites and ascitic fluid “humoral tests of malignancy”. J. Clin. Gastroenterol. 1994, 18, 94–8.
- Szturmowicz, M.; Tomkowski, W.; Fijalkowska, A.; Burakowski, J.; Sakowicz, A.; Filipecki, S. The role of carcinoembryonic antigen (CEA) and neuron-specific enolase (NSE) evaluation in pericardial fluid for the recognition of malignant pericarditis. Int. J. Biol. Markers 12, 96–101.
- Blake, G. J.; Ridker, P. M. Inflammatory bio-markers and cardiovascular risk prediction. J. Intern. Med. 2002, 252, 283–94.
- Sipe, J. D. Acute-phase proteins in osteoarthritis. Semin. Arthritis Rheum. 1995, 25, 75–86.
- St Clair, E. W. Tides of inflammation: impact of biologics. J. Rheumatol. Suppl. 2002, 65, 22–6.
- Montón, C.; Torres, A. Lung inflammatory response in pneumonia. Monaldi Arch. chest Dis. = Arch. Monaldi per le Mal. del torace / Fond. Clin. del Lav. IRCCS [and] Ist. di Clin. Tisiol. e Mal. Appar. Respir. Univ. di Napoli, Second. ateneo 1998, 53, 56–63.
- Sadoshima, J.; Izumo, S. Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes: potential involvement of an autocrine/paracrine mechanism. EMBO J. 1993, 12, 1681–92.
- Yamazaki, T.; Komuro, I.; Yazaki, Y. Molecular mechanism of cardiac cellular hypertrophy by mechanical stress. J. Mol. Cell. Cardiol. 1995, 27, 133–40.
- Wu, M.; Frieboes, H. B.; McDougall, S. R.; Chaplain, M. A. J.; Cristini, V.; Lowengrub, J. The effect of interstitial pressure on tumor growth: coupling with the blood and lymphatic vascular systems. J. Theor. Biol. 2013, 320, 131–51.
- Fleury, V.; Schwartz, L. Diffusion limited aggregation from shear stress as a simple model of vasculogenesis. Fractals 1999, 7, 33–39.
- Alfarouk, K. O.; Ibrahim, M. E.; Gatenby, R. A.; Brown, J. S. Riparian ecosystems in human cancers. Evol. Appl. 2013, 6, 46–53.
- Chang, Y. S.; di Tomaso, E.; McDonald, D. M.; Jones, R.; Jain, R. K.; Munn, L. L. Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 14608–13.
- Danciu, T. E.; Gagari, E.; Adam, R. M.; Damoulis, P. D.; Freeman, M. R. Mechanical strain delivers anti-apoptotic and proliferative signals to gingival fibroblasts. J. Dent. Res. 2004, 83, 596–601.
- Wang, X.; Yu, W.; Zheng, L. The dynamics of NF-κB pathway regulated by circadian clock. Math. Biosci. 2015, 260, 47–53.
- Lardner, A. The effects of extracellular pH on immune function. J. Leukoc. Biol. 2001, 69, 522–30.
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V. P. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–95.
- Alfarouk, K. O.; Shayoub, M. E. A. A.; Muddathir, A. K.; Elhassan, G. O.; Bashir, A. H. H. H. Evolution of Tumor Metabolism might Reflect Carcinogenesis as a Reverse Evolution process (Dismantling of Multicellularity). Cancers (Basel). 2011, 3, 3002–3017.
- Padma, M. V; Said, S.; Jacobs, M.; Hwang, D. R.; Dunigan, K.; Satter, M.; Christian, B.; Ruppert, J.; Bernstein, T.; Kraus, G.; Mantil, J. C. Prediction of pathology and survival by FDG PET in gliomas. J. Neurooncol. 2003, 64, 227–37.
- Singh, D.; Miles, K. Multiparametric PET/CT in oncology. Cancer Imaging 2012, 12, 336–44.
- Elliott, R.; Barnett, B. Ultrastructural Observation of Mitochondria in Human Breast Carcinoma Cells. Microsc. Microanal. 2011, 17, 194–195.
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–36.
- Keizer, H. G.; Pinedo, H. M.; Schuurhuis, G. J.; Joenje, H. Doxorubicin (adriamycin): a critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol. Ther. 1990, 47, 219–31.
- Guénin, S.; Schwartz, L.; Morvan, D.; Steyaert, J. M.; Poignet, A.; Madelmont, J. C.; Demidem, A. PP2A activity is controlled by methylation and regulates oncoprotein expression in melanoma cells: a mechanism which participates in growth inhibition induced by chloroethylnitrosourea treatment. Int. J. Oncol. 2008, 32, 49–57.
- Liang, X.-J.; Finkel, T.; Shen, D.-W.; Yin, J.-J.; Aszalos, A.; Gottesman, M. M. SIRT1 contributes in part to cisplatin resistance in cancer cells by altering mitochondrial metabolism. Mol. Cancer Res. 2008, 6, 1499–506.
- Wallace, K. B. Adriamycin-induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc. Toxicol. 2007, 7, 101–7.
- Cullen, K. J.; Yang, Z.; Schumaker, L.; Guo, Z. Mitochondria as a critical target of the chemotheraputic agent cisplatin in head and neck cancer. J. Bioenerg. Biomembr. 2007, 39, 43–50.
- Matsuyama, S.; Xu, Q.; Velours, J.; Reed, J. C. The Mitochondrial F0F1-ATPase proton pump is required for function of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell 1998, 1, 327–36.
- Leduc, E. H.; Wilson, J. W. Effect of Essential Fatty Acid Deficiency on Ultrastructure and Growth of Transplantable Mouse Hepatoma BRL. J Natl Cancer Inst 1964, 33, 721–739.
- Henry-Mowatt, J.; Dive, C.; Martinou, J.-C.; James, D. Role of mitochondrial membrane permeabilization in apoptosis and cancer. Oncogene 2004, 23, 2850–60.
- Wang, P.; Xu, C.; Xia, X.; Xu, J.; Wang, X.; Xiang, J.; Leung, A. W. Mitochondrial damage in nasopharyngeal carcinoma cells induced by ultrasound radiation in the presence of hypocrellin B. J. Ultrasound Med. 2010, 29, 43–50.
- Michelakis, E. D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.-L.; Mackey, J. R.; Fulton, D.; Abdulkarim, B.; McMurtry, M. S.; Petruk, K. C. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34.
- Schwartz, L.; Abolhassani, M.; Guais, A.; Sanders, E.; Steyaert, J.-M.; Campion, F.; Israël, M. A combination of alpha lipoic acid and calcium hydroxycitrate is efficient against mouse cancer models: preliminary results. Oncol. Rep. 2010, 23, 1407–16.
- Schwartz, L.; Guais, A.; Israël, M.; Junod, B.; Steyaert, J.-M.; Crespi, E.; Baronzio, G.; Abolhassani, M. Tumor regression with a combination of drugs interfering with the tumor metabolism: efficacy of hydroxycitrate, lipoic acid and capsaicin. Invest. New Drugs 2013, 31, 256–64.
- Abolhassani, M.; Guais, A.; Sanders, E.; Campion, F.; Fichtner, I.; Bonte, J.; Baronzio, G.; Fiorentini, G.; Israël, M.; Schwartz, L. Screening of well-established drugs targeting cancer metabolism: reproducibility of the efficacy of a highly effective drug combination in mice. Invest. New Drugs 2012, 30, 1331–42.
- Zachar, Z.; Marecek, J.; Maturo, C.; Gupta, S.; Stuart, S. D.; Howell, K.; Schauble, A.; Lem, J.; Piramzadian, A.; Karnik, S.; Lee, K.; Rodriguez, R.; Shorr, R.; Bingham, P. M. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J. Mol. Med. (Berl). 2011, 89, 1137–48.
- Baronzio, G.; Schwartz, L.; Crespi, E.; Guais, A.; Sanders, E.; Delépine, N.; Fiorentini, G. Early clinical and toxicological results of a combination of natural glycolysis inhibitors (METABLOCTM) on cancer patients. Biomed. Res. 2012, 23, 219–222.
- Schwartz, L.; Buhler, L.; Icard, P.; Lincet, H.; Steyaert, J.-M. Metabolic treatment of cancer: intermediate results of a prospective case series. Anticancer Res. 2014, 34, 973–80.
- Schwartz, L.; Buhler, L.; Icard, P.; Lincet, H.; Summa, G. M.; Steyaert, J.-M. Metabolic cancer treatment: Intermediate results of a clinical study. Cancer Ther. 2014, 10, 13–19.
- Schwartz, L.; Gabillet, J.; Buhler, L.; Steyaert, J.-M. The addition of chloroquine and metformine to Metabloc induces a rapid drop of tumor markers in advanced carcinoma. Cancer Ther. 2014, 10, 20–27.
- Elliott, R. L.; Jiang, X. P.; Head, J. F. Mitochondria organelle transplantation: introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res. Treat. 2012, 136, 347–54.
- Kaipparettu, B. A.; Ma, Y.; Park, J. H.; Lee, T.-L.; Zhang, Y.; Yotnda, P.; Creighton, C. J.; Chan, W.-Y.; Wong, L.-J. C. Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways. PLoS One 2013, 8, e61747.
- Pino-Barrio, M. J.; García-García, E.; Menéndez, P.; Martínez-Serrano, A. V-myc immortalizes human neural stem cells in the absence of pluripotency-associated traits. PLoS One 2015, 10, e0118499.
- Page, S.; Salem, M.; Laughlin, M. R. Intracellular Mg2+ regulates ADP phosphorylation and adenine nucleotide synthesis in human erythrocytes. Am. J. Physiol. 1998, 274, E920-7.
Figure and Legend:
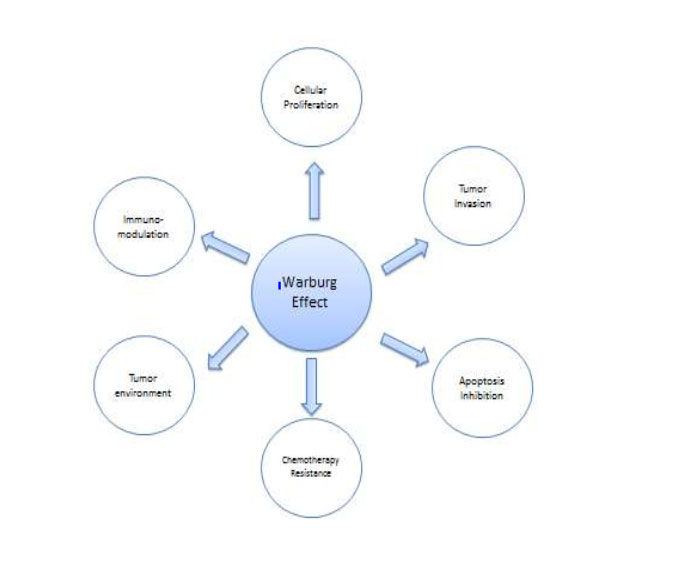 Figure1: show the possible correlation between Warburg Effect and Hallmarks of Cancer
Figure1: show the possible correlation between Warburg Effect and Hallmarks of Cancer
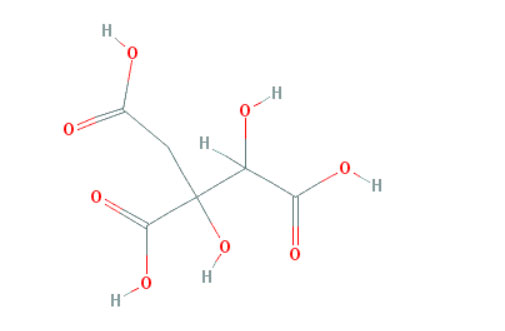
Figure 2: Shows the chemical structure of Hydroxycitric acid

Il y a des affirmations erronées ou imprécises dans cet article.
Comme par exemple le mode en fonctionnement énergétique anaérobie soit disant exclusif aux cellules cancéreuses, dans le monde sportif les chercheurs ont montré qu’en fonction du type de sport et d’intensité de l’effort à un moment les cellules musculaires en manque d’oxygène entreront en mode énergétique anaérobie [1], qui s’il dure trop longtemps entrainera la fatigue et des crampes par excédent d’acide lactique. Ces études permettent aux sportifs de haut niveau, par un entrainement adéquat assisté de mesure d’acidité lactique [2], d’éviter au mieux ce mode de fonctionnement pour améliorer leurs performances.
En fait la différence entre les cellules cancéreuses et les cellules normales, c’est que dans les cellules cancéreuses une part plus ou moins importantes des mitochondries ne fonctionnent plus qu’en mode anaérobie permanent ( une cellule contient de plusieurs centaines à plusieurs milliers de mitochondries, selon le type de cellules et leur sollicitation énergétique), Laurent CHATRE du CNRS et institut Pasteur a conçu l’appareil mTrip qui permet de visualiser l’état des mitochondries dans les cellules, de manière qualitative et quantitative [3].
D’autre part vous citez le régime cétogène en complément du métabloc pour son action restrictive sur l’alimentation des cellules cancéreuses, mais vous oubliez de citer le jeune intermittent validé par les recherches de Valter LONGO. il a démontré qu’un jeune à l’eau de 3 jours avec restriction calorique à 200kcal/jour permettrait de passer en cétose, mais surtout de régénérer les cellules souches des globules blanc en même temps qu’en éliminant les globules blancs endommagés et donc de régénérer le système immunitaire [4,5].
200kcal peut par exemple correspondre à 70g de spiruline/jour ou 30g de ketocal 4:1, mais vous pouvez faire aussi vos recettes à 200Kcal/jour vous même.
1. https://fr.wikipedia.org/wiki/M%C3%A9tabolisme_ana%C3%A9robie_lactique
2. https://www.newpharma.fr/search-results/premiers-soins-soins-a-domicile/autotests/1265-1352/2accutrend.html?gclid=CjwKCAiA07PRBRBJEiwAS20SIMR0FLud7e1meaqx-Al0WigNOGBDkhLB4tykqyLknNqFGQbnHGLf3RoCDI4QAvD_BwE&key=accutrend&s_kwcid=AL%21470%213%21225538766343%21p%21%21g%21%21accutrend&cid=paidsearch&ef_id=Wiv_KgAAAKadNXFi%3A20171210205241%3As
3. https://www.researchgate.net/publication/284670350_A_Single-Cell_Resolution_Imaging_Protocol_of_Mitochondrial_DNA_Dynamics_in_Physiopathology_mTRIP_Which_Also_Evaluates_Sublethal_Cytotoxicity
4. http://www.wikistrike.com/2017/05/des-scientifiques-decouvrent-que-le-jeune-declenche-la-regeneration-des-cellules-souches-et-combat-le-cancer.html
5. http://www.zoelho.com/ZoelhoFR/Publish/Regimes/Calorie-restrictie.htm
Bravo et merci !
Une « malade » qui se sent en forme 😉