 Peggy P.Hsu12David M.Sabatini123
Peggy P.Hsu12David M.Sabatini123
Disponible en ligne le 4 septembre 2008.Voir plus
Décrit il y a des décennies, l’effet Warburg de la glycolyse aérobie est une des principales caractéristiques métaboliques du cancer, mais son importance demeure incertaine. Dans cet essai, nous réexaminons l’effet de Warburg et établissons un cadre pour comprendre sa contribution à l’altération du métabolisme des cellules cancéreuses.
Texte principal
Il est difficile d’entamer une discussion sur le métabolisme des cellules cancéreuses sans d’abord mentionner Otto Warburg. Pionnier dans l’étude de la respiration, Warburg a fait une découverte marquante dans les années 1920. Il a découvert que, même en présence d’oxygène abondant, les cellules cancéreuses préfèrent métaboliser le glucose par glycolyse, ce qui semble paradoxal puisque la glycolyse, par rapport à la phosphorylation oxydative, est une voie moins efficace pour produire l'[tipso tip= »Adénosine-TriphosPhate »]ATP[/tipso] (Warburg, 1956). L’effet de Warburg a été démontré depuis lors dans différents types de tumeurs et l’augmentation concomitante de l’absorption de glucose a été exploitée cliniquement pour la détection de tumeurs par tomographie d’émission de positrons par fluorodésoxyglucose (FDG-PET). Bien que la glycolyse aérobie soit maintenant généralement reconnue comme une marque métabolique du cancer, son lien de causalité avec la progression du cancer n’est toujours pas clair. Dans cet essai, nous discutons des facteurs, des avantages et des responsabilités possibles de l’altération du métabolisme des cellules cancéreuses (figure 1). Bien que notre accent sur l’effet de Warburg reflète l’orientation du domaine, nous aimerions également encourager une approche plus large de l’étude du métabolisme du cancer qui tienne compte des contributions de toutes les petites voies moléculaires interconnectées de la cellule.
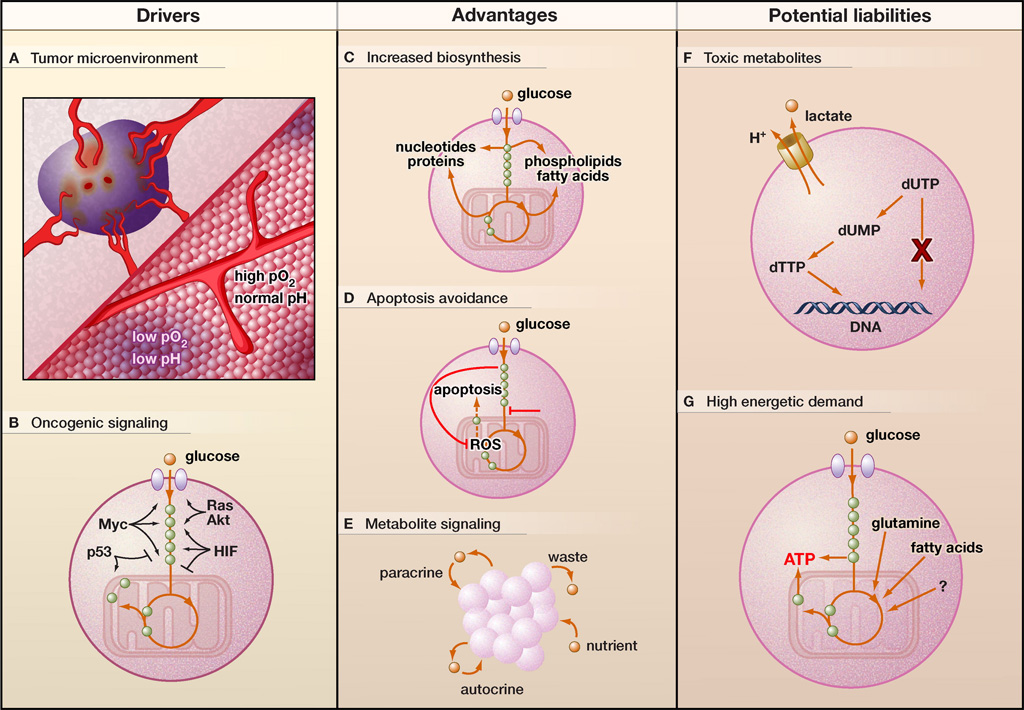 Figure 1. Le métabolisme altéré des cellules cancéreuses
Figure 1. Le métabolisme altéré des cellules cancéreuses
Conducteurs (A et B). Les dérangements métaboliques dans les cellules cancéreuses peuvent provenir soit de la sélection de cellules adaptées au micro environnement tumoral, soit d’une signalisation aberrante due à l’activation oncogène. Le micro environnement tumoral est hétérogène dans l’espace et dans le temps, contenant des régions à faible teneur en oxygène et à faible pH (violet). De plus, de nombreuses voies cancéreuses de signalisation associées au cancer induisent une reprogrammation métabolique. Les gènes cibles activés par le facteur inductible par l’hypoxie ([tipso tip= »Hypoxia Inducible Factors »]HIF[/tipso]) diminuent la dépendance de la cellule à l’oxygène, tandis que [tipso tip= »Protéine codée par le protooncogène c-ras »]Ras[/tipso], [tipso tip= »Myc est un proto-oncogène qui est sur-exprimé dans certains cancers humains. Quand il est soumis à des mutations ou à une sur-expression, il stimule la prolifération des cellules et se conduit comme un oncogène. »]Myc[/tipso] et [tipso tip= »Protéine Kinase B (PKB) est une protéine essentielle dans la signalisation des cellules des mammifères. »]Akt[/tipso] peuvent également réguler la consommation de glucose et la glycolyse. La perte de [tipso tip= »Le p53 est un facteur de transcription régulant de multiples fonctions cellulaires importantes comme la régulation du cycle cellulaire, l’autophagie ou l’apoptose. Chez l’homme, ce gène est situé sur le chromosome 17″]p53[/tipso] peut également récapituler les caractéristiques de l’effet de Warburg, à savoir le découplage de la glycolyse des niveaux d’oxygène.
Avantages (C-E). L’altération du métabolisme des cellules cancéreuses est susceptible de leur conférer plusieurs avantages prolifératifs et de survie, tels que la possibilité pour les cellules cancéreuses d’effectuer la biosynthèse de macromolécules (C), d’éviter l’apoptose (D) et de s’engager dans la signalisation paracrine et autocrine locale basée sur le métabolite (E).
Passifs éventuels (F et G). Toutefois, cette altération du métabolisme peut également conférer plusieurs vulnérabilités aux cellules cancéreuses. Par exemple, un métabolisme régulé à la hausse peut entraîner l’accumulation de métabolites toxiques, y compris le lactate et les nucléotides non canoniques, qui doivent être éliminés (F). De plus, les cellules cancéreuses peuvent également présenter une forte demande énergétique, pour laquelle elles doivent soit augmenter le flux par des processus générateurs d’ATP normaux, soit compter sur une plus grande diversité de sources d’énergie (G).
Le Micro-environnement Tumoral choisit un Métabolisme Altéré
Une idée force pour expliquer l’effet de Warburg est que le métabolisme altéré des cellules cancéreuses confère un avantage sélectif pour la survie et la prolifération dans le micro-environnement tumoral unique. Au fur et à mesure que la tumeur se développe, elle dépasse les limites de diffusion de son apport sanguin local, ce qui entraîne une hypoxie et la stabilisation du facteur de transcription inductible par l’hypoxie, HIF. HIF lance un programme transcriptionnel qui fournit de multiples solutions au stress hypoxique (révisé dans Kaelin et Ratcliffe, 2008). Parce qu’une diminution de la dépendance à la respiration aérobie devient avantageuse, le métabolisme cellulaire est déplacé vers la glycolyse par l’expression accrue des enzymes glycolytiques, des transporteurs de glucose et des inhibiteurs du métabolisme mitochondrial. En outre, HIF stimule l’angiogenèse (la formation de nouveaux vaisseaux sanguins) en régulant plusieurs facteurs, dont le facteur de croissance endothéliale vasculaire (VEGF) le plus proéminent.
Cependant, les vaisseaux sanguins recrutés dans le micro-environnement tumoral sont désorganisés, peuvent ne pas livrer le sang de façon efficace et, par conséquent, n’atténuent pas complètement l’hypoxie (voir Gatenby et Gillies, 2004). Les niveaux d’oxygène dans une tumeur varient à la fois spatialement et temporellement, et les cycles résultants des niveaux fluctuants d’oxygène peuvent potentiellement sélectionner pour les tumeurs qui régulent constitutionnellement la glycolyse. Il est intéressant de noter qu’à l’exception peut-être des tumeurs qui ont perdu la protéine von Hippel-Lindau (VHL), qui est normalement la médiatrice de la dégradation de l’HIF, l’HIF est toujours couplée aux niveaux d’oxygène, comme en témoigne l’hétérogénéité de l’expression de l’HIF dans le micro-environnement tumoral (Wiesener et al., 2001; Zhong et al., 1999). Par conséquent, l’effet de Warburg – c’est-à-dire un découplage de la glycolyse des niveaux d’oxygène – ne peut pas s’expliquer uniquement par une augmentation de la régulation du HIF. D’autres mécanismes moléculaires sont susceptibles d’être importants, tels que les changements métaboliques induits par l’activation oncogène et la perte de suppresseurs tumoraux.
L’activation d’oncogène entraîne des changements dans le métabolisme
Non seulement le micro-environnement tumoral peut-il être sélectionné pour un métabolisme dérangé, mais le statut oncogène peut également entraîner des changements métaboliques. Depuis l’époque de Warburg, l’étude biochimique du métabolisme du cancer a été éclipsée par les efforts visant à identifier les mutations qui contribuent à l’initiation et à la progression du cancer. Des travaux récents ont toutefois démontré que les composants clés de l’effet de Warburg – augmentation de la consommation de glucose, diminution de la phosphorylation oxydative et production de lactate qui l’accompagne – sont également des caractéristiques distinctives de l’activation oncogène. La molécule de signalisation Ras, oncogène puissant en cas de mutation, favorise la glycolyse (révisée dans Dang et Semenza, 1999; Ramanathan et al., 2005). L’akt kinase, un effecteur en aval bien caractérisé de la signalisation de l’insuline, reprend son rôle dans l’absorption et l’utilisation du glucose dans le contexte du cancer (révisé dans Manning et Cantley, 2007), tandis que le facteur de transcription de Myc régule l’expression de divers gènes métaboliques (révisé dans Gordan et al., 2007). La voie la plus parcimonieuse vers la tumorigénèse peut être l’activation de nœuds oncogènes clés qui exécutent un programme prolifératif, dont le métabolisme peut être un bras important. De plus, la régulation du métabolisme n’est pas exclusive aux oncogènes. La perte de la protéine suppresseur de tumeur p53 empêche l’expression du gène codant pour SCO2 (la synthèse de la protéine cytochrome c oxydase), qui interfère avec le fonctionnement de la chaîne respiratoire mitochondriale (Matoba et al., 2006). Un deuxième effecteur p53, le TIGAR (régulateur de glycolyse et d’apoptose induit par PT53), inhibe la glycolyse en diminuant les niveaux de fructose-2,6-bisphosphate, un puissant stimulateur de glycolyse et inhibiteur de la gluconéogenèse (Bensaad et al., 2006). D’autres travaux suggèrent également que la régulation du métabolisme du glucose par voie p53 pourrait dépendre du facteur de transcription NF-KB (Kawauchi et al., 2008).
Il a été démontré que l’inhibition de la lactate déshydrogénase A (LDH-A) empêche l’effet de Warburg et force les cellules cancéreuses à retourner à la phosphorylation oxydante afin de réoxyder la NADH et de produire de l’ATP (Fantin et al., 2006; Shim et al., 1997). Bien que les cellules soient aptes à la respiration, elles présentent une croissance tumorale atténuée, ce qui suggère que la glycolyse aérobie pourrait être essentielle à la progression du cancer. Dans un modèle de culture cellulaire de fibroblastes primaires à transformation maligne progressive par surexpression de la télomérase, de l’antigène T petit et grand, et de l’oncogène H-Ras, la tumorigénicité croissante est corrélée avec la sensibilité à l’inhibition glycolytique. Cette constatation suggère que l’effet de Warburg pourrait être inhérent aux événements moléculaires de transformation (Ramanathan et al., 2005). Cependant, l’introduction de facteurs définis similaires dans les cellules souches mésenchymateuses (MSC) humaines a révélé que la transformation peut être associée à une dépendance accrue à la phosphorylation oxydative (Funes et al., 2007). Il est intéressant de noter que, lorsqu’ils sont introduits in vivo, ces MSC transformés régulent les gènes glycolytiques, un effet qui est inversé lorsque les cellules sont explosées et mises en culture dans des conditions normoxiques. Ces modèles contrastés suggèrent que l’effet de Warburg peut dépendre du contexte, dans certains cas, de changements génétiques et dans d’autres, des exigences du microenvironnement. Indépendamment du fait que le microenvironnement tumoral ou l’activation oncogène joue un rôle plus important dans le développement d’un métabolisme cancéreux distinct, il est probable que les modifications qui en résultent confèrent des avantages adaptatifs, prolifératifs et de survie à la cellule cancéreuse.
Le Métabolisme Altéré fournit des Substrats pour les Voies Biosynthétiques
Bien que les études sur le métabolisme du cancer aient été largement centrées sur l’énergie, les cellules à division rapide ont des besoins variés. Les cellules de prolifération ont besoin non seulement d’ATP mais aussi de nucléotides, d’acides gras, de lipides membranaires et de protéines, et un métabolisme reprogrammé peut servir à soutenir la synthèse des macromolécules. Des études récentes ont montré que plusieurs étapes de la synthèse lipidique sont nécessaires et pourraient même promouvoir activement la tumorigénèse. L’inhibition de la citrate lyase ATP, l’enzyme distale qui convertit le citrate dérivé des mitochondries en acétylcoenzyme A cytosolique, précurseur de nombreuses espèces de lipides, prévient la prolifération des cellules cancéreuses et la croissance tumorale (Hatzivassiliou et al., 2005). La synthase d’acide gras synthétisé, exprimée à de faibles concentrations dans les tissus normaux, est régulée à la hausse dans le cancer et peut également être nécessaire pour la tumorigénèse (révisée dans Menendez et Lupu, 2007). De plus, les cellules cancéreuses peuvent également améliorer leurs capacités biosynthétiques en exprimant une forme tumorale spécifique de pyruvate kinase (PK), M2-PK. La pyruvate kinase catalyse la troisième réaction irréversible de glycolyse, la conversion du phosphoénolpyruvate (PEP) en pyruvate. Étonnamment, on pense que le M2-PK des cellules cancéreuses est moins actif dans la conversion du PEP en pyruvate et donc moins efficace à la production d’ATP (révisé dans Mazurek et al., 2005). Un avantage majeur pour la cellule cancéreuse, cependant, est que les intermédiaires glycolytiques en amont de la PEP pourraient être réorientés vers des processus synthétiques. Des travaux récents ont révélé que le M2-PK spécifique au cancer entraîne une augmentation de l’incorporation des glucides dans les lipides et, en élargissant le lien entre la signalisation des facteurs de croissance et le métabolisme du cancer, peut être régulé par la fixation de la phosphotyrosine (Christofk et al., 2008a, 2008b).
Toutefois, la fabrication des éléments constitutifs de la cellule entraîne un coût énergétique et ne peut pas expliquer entièrement l’effet de Warburg. La biosynthèse, en plus d’entraîner une augmentation inhérente de la demande d’ATP pour exécuter des réactions synthétiques, devrait également entraîner une diminution de l’offre d’ATP à mesure que divers intermédiaires glycolytiques et intermédiaires du cycle de Krebs sont détournés. La synthèse lipidique, par exemple, nécessite la coopération de la glycolyse, du cycle de Krebs et du shunt de phosphate de pentose. Comme le pyruvate doit entrer dans les mitochondries dans ce cas, il évite la conversion en lactate et ne peut donc pas contribuer à l’ATP dérivé de la glycolyse. De plus, alors qu’une biosynthèse accrue peut expliquer la faim de glucose des cellules cancéreuses, elle ne peut expliquer l’augmentation de la production d’acide lactique décrite à l’origine par Warburg, ce qui suggère que le lactate doit également résulter du métabolisme des substrats non glucosés. Récemment, il a été démontré que la glutamine peut être métabolisée par le cycle de l’acide citrique dans les cellules cancéreuses et transformée en lactate, ce qui produit du NADPH pour la biosynthèse des lipides et de l’oxaloacetate pour la reconstitution des intermédiaires du cycle de Krebs (DeBerardinis et al., 2007).
Les Voies Métaboliques Régulent l’Apoptose
En plus de l’implication dans la prolifération, un métabolisme altéré peut favoriser une autre fonction essentielle au cancer: la prévention de l’apoptose. La perte de la cible p53 TIGAR sensibilise les cellules cancéreuses à l’apoptose, très probablement en provoquant une augmentation des espèces réactives d’oxygène (Bensaad et al., 2006). D’autre part, la surexpression de la glycéroldéhyde-3-phosphate déshydrogénase (GAPDH) empêche la mort cellulaire indépendante de la caspase, probablement en stimulant la glycolyse, en augmentant les niveaux d’ATP cellulaire et en favorisant l’autophagie (Colell et al., 2007). Il reste à déterminer si le GAPDH joue ou non un rôle physiologique dans la régulation de la mort cellulaire.
De façon intrigante, Bonnet et al. (2007) ont rapporté que le traitement des cellules cancéreuses par l’acétate de dichloro (Dichloroacétate (DCA), un petit inhibiteur de la pyruvate déshydrogénase kinase, a des effets frappants sur leur survie et sur la croissance des tumeurs xénogreffées. Le DCA, un traitement actuellement approuvé contre l’acidose lactique congénitale, active la phosphorylation oxydative et favorise l’apoptose par deux mécanismes. Premièrement, l’augmentation du flux à travers la chaîne de transport des électrons provoque la dépolarisation du potentiel de la membrane mitochondriale (que les auteurs ont trouvé hyperpolarisé spécifiquement dans les cellules cancéreuses) et la libération du cytochrome effecteur apoptotique c. Deuxièmement, une augmentation des espèces réactives d’oxygène générées par phosphorylation oxydative régule le canal K+ sous tension, conduisant à l’efflux d’ions potassiques et à l’activation de la caspase. Leurs travaux suggèrent que les cellules cancéreuses peuvent déplacer leur métabolisme vers la glycolyse afin de prévenir la mort cellulaire et que le fait de forcer les cellules cancéreuses à respirer par voie aérobie peut contrecarrer cette adaptation. Bien que ce travail préliminaire ait incité certains patients atteints de cancer à l’automédication avec le DCA, un essai clinique contrôlé sera essentiel pour démontrer sans équivoque l’innocuité et l’efficacité du DCA en tant qu’agent anticancéreux.
Les Cellules Cancéreuses Peuvent Signaler Localement dans le Micro-environnement Tumoral
Les cellules cancéreuses peuvent recâbler les voies métaboliques pour exploiter le micro-environnement tumoral et favoriser la signalisation spécifique au cancer. Sans l’accès à la circulation centrale, il est possible que les métabolites puissent être concentrés localement et atteindre des niveaux suprasystémiques, ce qui permet aux cellules cancéreuses de s’engager dans la signalisation autocrine et paracrine à médiation métabolite qui ne se produit pas dans les tissus normaux. Les cancers de la prostate dits androgènes indépendants de l’androgène ne peuvent être indépendants que des androgènes exogènes synthétisés surrénales. Les cellules du cancer de la prostate androgénétiques indépendantes de l’androgène expriment encore le récepteur androgénique et peuvent être capables de synthétiser leurs propres androgènes de façon autonome (Stanbrough et al., 2006).
Peut-être l’idée la plus provocante, mais pas encore éprouvée, est que les métabolites dans le microenvironnement tumoral à diffusion limitée pourraient agir comme molécules de signalisation paracrine. Traditionnellement considéré comme un déchet glycolytique, le lactate peut être un de ces signaux. Comme nous l’avons mentionné plus haut, on a constaté que l’inhibition de la déshydrogénase du lactate peut bloquer la croissance tumorale, très probablement par de multiples mécanismes. Une grande partie des données probantes sur le lactate en tant que métabolite multifonctionnel proviennent de travaux sur la physiologie de l’exercice et le métabolisme musculaire (examinés dans Philp et al., 2005). Transporté par plusieurs transporteurs de monocarboxylates, le lactate peut être partagé et métabolisé entre les cellules, bien que l’idée soit encore controversée (Hashimoto et al., 2006; Yoshida et al., 2007). L’interconversion du lactate et du pyruvate pourrait modifier le rapport NAD+/NADH dans les cellules, et l’échange de lactate pourrait servir à coordonner le métabolisme d’un groupe de cellules. L’interaction tumeur-stroma peut donc avoir une composante métabolique (Koukourakis et al., 2006). Les cellules cancéreuses réagissent de façon autonome à l’hypoxie pour déclencher l’angiogenèse, et il serait donc passionnantqu’un métabolite comme le lactate puisse amplifier positivement ce programme angiogénique, un processus qui nécessite un effort semi-coordonné entre plusieurs cellules. En effet, l’acidose précède souvent l’angiogenèse, et le lactate peut stimuler l’expression HIF indépendamment de l’hypoxie (Fukumura et al., 2001; Lu et al., 2002; Shi et al., 2001). Les cellules cancéreuses, en participant à une sorte de quorum et en coordonnant leur métabolisme, peuvent donc agir comme un pseudo-organe.
Le métabolisme comme modulateur en amont des voies de signalisation
Non seulement le métabolisme est-il en aval des voies oncogènes, mais un métabolisme en amont modifié peut affecter l’activité des voies de signalisation qui détectent normalement l’état de la cellule. Les personnes présentant des mutations héréditaires de la succinate déshydrogénase et de la fumarate hydratase développent des tumeurs hautement angiogéniques, tout comme celles qui présentent une perte de la protéine suppresseur de la tumeur VHL qui agit en amont de l’HIF (revue dans Kaelin et Ratcliffe, 2008). Le mécanisme de tumorigénèse dans ces syndromes cancéreux est encore controversé. Cependant, il a été proposé que la perte de succinate déshydrogénase et de fumarate hydratase cause une accumulation de succinate ou de fumarate, respectivement, conduisant à l’inhibition des prolylhydroxyases qui marquent HIF pour la dégradation provoquée par le VHL (Isaacs et al., 2005; Pollard et al., 2005; Selak et al., 2005). Dans ce cas rare, la succinate déshydrogénase et la fumarate hydratase agissent comme de véritables suppresseurs de tumeurs.
Les mutations dans les gènes métaboliques, cependant, n’ont pas besoin d’être un événement cancérigène. Plus subtilement, l’activation de diverses voies métaboliques pourrait moduler l’activité des facteurs pro-cancéreux en aval. Alors qu’il est bien admis que la signalisation des facteurs de croissance est généralement dérégulée dans le cancer, l’implication des éléments nutritifs ou de la signalisation énergétique dans le cancer demeure incertaine. Dans les procaryotes, divers métabolites sont détectés directement par la machine de signalisation. Les voies de migration des mammifères qui réagissent à l’état énergétique et nutritionnel peuvent aussi interagir directement avec les métabolites. Il est bien établi que l’AMP-kinase détecte le rapport AMP/ATP (révisé dans Hardie, 2007), tandis que le mTOR (la cible mammalienne de la rapamycine) détecte les concentrations d’acides aminés cellulaires (Kim et al., 2008; Sancak et al., 2008). L’AMP-kinase et le mTOR ont tous deux été associés à des syndromes tumoraux. Il est possible qu’une façon de réguler ces voies de signalisation favorables à la croissance consiste à augmenter les niveaux des métabolites normaux qu’ils perçoivent.
La Régulation du Métabolisme Génère des Sous-produits Toxiques
Bien qu’une altération du métabolisme confère plusieurs avantages à la cellule cancéreuse, elle n’est pas sans inconvénients. À la suite d’un métabolisme dérangé ou tout simplement hyperactif, les cellules cancéreuses peuvent être chargées de sous-produits toxiques qui doivent être éliminés. Jusqu’ à présent, il y a relativement peu de preuves de cette hypothèse dans la littérature existante, mais quelques exemples suggèrent que les cellules cancéreuses ont besoin de mécanismes de désintoxication pour maintenir leur survie. Bien qu’il existe des enzymes qui détoxiquent les toxines exogènes, plusieurs enzymes de « nettoyage domestique », un terme inventé à partir d’études sur des bactéries, traitent des métabolites toxiques endogènes (revues dans Galperin et al., 2006). Les meilleurs exemples d’enzymes de « nettoyage domestique » sont les hydrolases NUDIX (diphosphate nucléosidique non canonique lié à un autre fragment X), une famille d’enzymes qui agissent sur le pool nucléotidique et éliminent les triphosphates nucléosidiques non canoniques. Lorsqu’ils sont incorporés dans l’ADN, ces nucléotides aberrants peuvent entraîner des inadéquations, des mutations et éventuellement la mort cellulaire. La dUTP pyrophosphatase (DUTP), qui hydrolyse le dUTP en dUMP et empêche l’incorporation des uraciles dans l’ADN, peut jouer un rôle dans la résistance aux inhibiteurs de la thymidylate synthase. La suppression du DUT sensibilise certaines cellules cancéreuses aux antimétabolites de la pyrimidine, ce qui suggère que l’inhibition de ces enzymes cellulaires de nettoyage domestique pourrait être une stratégie chimiothérapeutique d’appoint efficace (Koehler et Ladner, 2004).
On pense que la production de lactate associée au passage à un métabolisme glycolytique contribue à l’acidification du microenvironnement. Il a été démontré que les cellules cancéreuses capables de s’adapter à un environnement acide et même d’en tirer parti, peuvent réguler à la hausse les H+-ATPases vacuolaires, les antiporteurs Na+-H+ et les transporteurs monocarboxylates liés à l’ H+ (revues dans Gatenby et Gillies, 2004). L’inhibition de ces mécanismes adaptatifs peut mener à une diminution de la viabilité des cellules cancéreuses et à une sensibilité accrue aux agents chimiothérapeutiques (voir Fais et coll., 2007; Fang et coll., 2006).
Territoire Inexploré
De nombreux mystères demeurent non élucidés dans notre compréhension du métabolisme humain, même normal, sans parler de celui des cellules cancéreuses. Les voies métaboliques de la cellule mammifère et leurs nombreuses interconnexions sont incomplètes, car de nombreuses enzymes ne sont pas encore notées dans le génome humain. Bien que nous ayons des suppositions par homologie, les identités des enzymes humaines qui catalysent les réactions que nous savons devoir se produire sont encore insaisissables. En plus d’annoter tous les gènes métaboliques humains, les » ins » et les » outs » (c. -à-d. les métabolites qui entrent et sortent des cellules) devraient être mesurés et catalogués. On ne sait pas non plus exactement quel pourcentage du combustible cellulaire est normalement utilisé pour la production d’ATP, la biosynthèse ou d’autres procédés. Et à quelques rares exceptions près, on en sait étonnamment peu sur le métabolisme intercellulaire. Une grande partie de notre compréhension du métabolisme a été héritée du travail dans des organismes simples; la nature compartimentée du métabolisme humain est un domaine passionnant d’exploration potentielle.
Bien que l’aérobie glycolyse soit le phénomène métabolique le plus caractérisé, quoique encore déroutant, dans le cancer, de nombreux autres aspects du métabolisme du cancer sont susceptibles d’être des dérèglements du métabolisme normal et devraient être élucidés. L’état nutritionnel du microenvironnement tumoral n’ a pas encore été examiné avec soin. Les cellules cancéreuses, bien qu’elles soient engagées dans des processus à faible consommation d’énergie, doivent néanmoins être en mesure de maintenir les niveaux d’ATP, soit en se fiant à un flux accru par glycolyse, soit en utilisant diverses sources de combustible. Plusieurs hypothèses existent quant à la raison pour laquelle une fraction des tumeurs sont réfractaires à l’imagerie par le FDG-PET. Une possibilité est que certaines cellules cancéreuses ne soient pas principalement des métaboliseurs de glucose, mais qu’elles puissent s’appuyer sur des sources de combustibles de remplacement, dont la caractérisation détaillée peut mener à la détection et au traitement de tumeurs « TEP-négatives ». En outre, il y a des questions plus complexes auxquelles il faut répondre: Est-il possible que les cellules cancéreuses présentent une « dépendance métabolique »? Existe-t-il des voies métaboliques uniques propres au cancer, ou des combinaisons de voies métaboliques, utilisées par la cellule cancéreuse mais pas par les cellules normales? Les différentes étapes de l’adaptation métabolique sont-elles nécessaires pour que la cellule cancéreuse progresse du stade de tumeur primaire à l’invasion et à la métastase? Le métabolisme du cancer est-il malléable?
D’un point de vue thérapeutique, la connaissance des causes, des bienfaits et des vulnérabilités du métabolisme des cellules cancéreuses permettra d’identifier de nouvelles cibles médicamenteuses et facilitera la conception de mimétiques métaboliques qui sont uniquement assimilés par les cellules cancéreuses ou transformés en forme active par des enzymes régulées dans les tumeurs. Le profilage de métabolites ou d’activités enzymatiques peut nous permettre de mettre au point des tests diagnostiques du cancer, et les dérivés métabolites peuvent être utilisés pour l’imagerie moléculaire du cancer, comme l’illustre le FDG-PET. Nous trouvons la possibilité d’une nouvelle classe de thérapies contre le cancer et d’outils diagnostiques particulièrement intéressants. Par conséquent, nous insistons sur la nécessité d’explorer au-delà d’un modèle de métabolisme du cancer axé sur le glucose et l’énergie pour en arriver à un modèle plus large qui englobe tous les besoins métaboliques d’une cellule cancéreuse. Il est peut-être temps de sortir de l’ombre de Warburg.
Remerciements
Nous remercions T. DiCesare pour son aide avec la figurine.
Références
- Bensaad et al., 2006
-
K. Bensaad, A. Tsuruta, M.A. Selak, M.N. Vidal, K. Nakano, R. Bartrons, E. Gottlieb, K.H. VousdenCell, 126 (2006), pp. 107-120
- Bonnet et al., 2007
-
S. Bonnet, S.L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, C.T. Lee, G.D. Lopaschuk, L. Puttagunta, G. Harry, et al.Cancer Cell, 11 (2007), pp. 37-51
- Christofk et al., 2008a
-
H.R. Christofk, M.G. Vander Heiden, M.H. Harris, A. Ramanathan, R.E. Gerszten, R. Wei, M.D. Fleming, S.L. Schreiber, L.C. CantleyNature, 452 (2008), pp. 230-233
- Christofk et al., 2008b
-
H.R. Christofk, M.G. Vander Heiden, N. Wu, J.M. Asara, L.C. CantleyNature, 452 (2008), pp. 181-186
- Colell et al., 2007
-
A. Colell, J.E. Ricci, S. Tait, S. Milasta, U. Maurer, L. Bouchier-Hayes, P. Fitzgerald, A. Guio-Carrion, N.J. Waterhouse, C.W. Li, et al.Cell, 129 (2007), pp. 983-997
- Dang and Semenza, 1999
-
C.V. Dang, G.L. SemenzaTrends Biochem. Sci., 24 (1999), pp. 68-72
- DeBerardinis et al., 2007
-
R.J. DeBerardinis, A. Mancuso, E. Daikhin, I. Nissim, M. Yudkoff, S. Wehrli, C.B. ThompsonProc. Natl. Acad. Sci. USA, 104 (2007), pp. 19345-19350
- Fais et al., 2007
-
S. Fais, A. De Milito, H. You, W. QinCancer Res., 67 (2007), pp. 10627-10630
- Fang et al., 2006
-
J. Fang, Q.J. Quinones, T.L. Holman, M.J. Morowitz, Q. Wang, H. Zhao, F. Sivo, J.M. Maris, M.L. WahlMol. Pharmacol., 70 (2006), pp. 2108-2115
- Fantin et al., 2006
-
V.R. Fantin, J. St-Pierre, P. LederCancer Cell, 9 (2006), pp. 425-434
- Fukumura et al., 2001
-
D. Fukumura, L. Xu, Y. Chen, T. Gohongi, B. Seed, R.K. JainCancer Res., 61 (2001), pp. 6020-6024
- Funes et al., 2007
-
J.M. Funes, M. Quintero, S. Henderson, D. Martinez, U. Qureshi, C. Westwood, M.O. Clements, D. Bourboulia, R.B. Pedley, S. Moncada, C. BoshoffProc. Natl. Acad. Sci. USA, 104 (2007), pp. 6223-6228
- Galperin et al., 2006
-
M.Y. Galperin, O.V. Moroz, K.S. Wilson, A.G. MurzinMol. Microbiol., 59 (2006), pp. 5-19
- Gatenby and Gillies, 2004
-
R.A. Gatenby, R.J. GilliesNat. Rev. Cancer, 4 (2004), pp. 891-899
- Gordan et al., 2007
-
J.D. Gordan, C.B. Thompson, M.C. SimonCancer Cell, 12 (2007), pp. 108-113
- Hardie, 2007
-
D.G. HardieNat. Rev. Mol. Cell Biol., 8 (2007), pp. 774-785
- Hashimoto et al., 2006
-
T. Hashimoto, R. Hussien, G.A. BrooksAm. J. Physiol. Endocrinol. Metab., 290 (2006), pp. E1237-E1244
- Hatzivassiliou et al., 2005
-
G. Hatzivassiliou, F. Zhao, D.E. Bauer, C. Andreadis, A.N. Shaw, D. Dhanak, S.R. Hingorani, D.A. Tuveson, C.B. ThompsonCancer Cell, 8 (2005), pp. 311-321
- Isaacs et al., 2005
-
J.S. Isaacs, Y.J. Jung, D.R. Mole, S. Lee, C. Torres-Cabala, Y.L. Chung, M. Merino, J. Trepel, B. Zbar, J. Toro, et al.Cancer Cell, 8 (2005), pp. 143-153
- Kaelin and Ratcliffe, 2008
-
W.G. Kaelin Jr., P.J. RatcliffeMol. Cell, 30 (2008), pp. 393-402
- Kawauchi et al., 2008
-
K. Kawauchi, K. Araki, K. Tobiume, N. TanakaNat. Cell Biol., 10 (2008), pp. 611-618
- Kim et al., 2008
-
E. Kim, P. Goraksha-Hicks, L. Li, T.P. Neufeld, K.L. GuanNat. Cell Biol., 10 (2008), pp. 935-945
- Koehler and Ladner, 2004
-
S.E. Koehler, R.D. LadnerMol. Pharmacol., 66 (2004), pp. 620-626
- Koukourakis et al., 2006
-
M.I. Koukourakis, A. Giatromanolaki, A.L. Harris, E. SivridisCancer Res., 66 (2006), pp. 632-637
- Lu et al., 2002
-
H. Lu, R.A. Forbes, A. VermaJ. Biol. Chem., 277 (2002), pp. 23111-23115
- Manning and Cantley, 2007
-
B.D. Manning, L.C. CantleyCell, 129 (2007), pp. 1261-1274
- Matoba et al., 2006
-
S. Matoba, J.G. Kang, W.D. Patino, A. Wragg, M. Boehm, O. Gavrilova, P.J. Hurley, F. Bunz, P.M. HwangScience, 312 (2006), pp. 1650-1653
- Mazurek et al., 2005
-
S. Mazurek, C.B. Boschek, F. Hugo, E. EigenbrodtSemin. Cancer Biol., 15 (2005), pp. 300-308
- Menendez and Lupu, 2007
-
J.A. Menendez, R. LupuNat. Rev. Cancer, 7 (2007), pp. 763-777
- Philp et al., 2005
-
A. Philp, A.L. Macdonald, P.W. WattJ. Exp. Biol., 208 (2005), pp. 4561-4575
- Pollard et al., 2005
-
P.J. Pollard, J.J. Briere, N.A. Alam, J. Barwell, E. Barclay, N.C. Wortham, T. Hunt, M. Mitchell, S. Olpin, S.J. Moat, et al.Hum. Mol. Genet., 14 (2005), pp. 2231-2239
- Ramanathan et al., 2005
-
A. Ramanathan, C. Wang, S.L. SchreiberProc. Natl. Acad. Sci. USA, 102 (2005), pp. 5992-5997
- Sancak et al., 2008
-
Y. Sancak, T.R. Peterson, Y.D. Shaul, R.A. Lindquist, C.C. Thoreen, L. Bar-Peled, D.M. SabatiniScience, 320 (2008), pp. 1496-1501
- Selak et al., 2005
-
M.A. Selak, S.M. Armour, E.D. MacKenzie, H. Boulahbel, D.G. Watson, K.D. Mansfield, Y. Pan, M.C. Simon, C.B. Thompson, E. GottliebCancer Cell, 7 (2005), pp. 77-85
- Shi et al., 2001
-
Q. Shi, X. Le, B. Wang, J.L. Abbruzzese, Q. Xiong, Y. He, K. XieOncogene, 20 (2001), pp. 3751-3756
- Shim et al., 1997
-
H. Shim, C. Dolde, B.C. Lewis, C.S. Wu, G. Dang, R.A. Jungmann, R. Dalla-Favera, C.V. DangProc. Natl. Acad. Sci. USA, 94 (1997), pp. 6658-6663
- Stanbrough et al., 2006
-
M. Stanbrough, G.J. Bubley, K. Ross, T.R. Golub, M.A. Rubin, T.M. Penning, P.G. Febbo, S.P. BalkCancer Res., 66 (2006), pp. 2815-2825
- Warburg, 1956
-
O. WarburgScience, 124 (1956), pp. 269-270
- Wiesener et al., 2001
-
M.S. Wiesener, P.M. Munchenhagen, I. Berger, N.V. Morgan, J. Roigas, A. Schwiertz, J.S. Jurgensen, G. Gruber, P.H. Maxwell, S.A. Loning, et al.Cancer Res., 61 (2001), pp. 5215-5222
- Yoshida et al., 2007
-
Y. Yoshida, G.P. Holloway, V. Ljubicic, H. Hatta, L.L. Spriet, D.A. Hood, A. BonenJ. Physiol., 582 (2007), pp. 1317-1335
- Zhong et al., 1999
-
H. Zhong, A.M. De Marzo, E. Laughner, M. Lim, D.A. Hilton, D. Zagzag, P. Buechler, W.B. Isaacs, G.L. Semenza, J.W. SimonsCancer Res., 59 (1999), pp. 5830-5835


L’ombre bien évidemment !
Très intéressant, mais en attendant que tous les mystères de la cellule cancéreuse soient percés, les malades ont tout intérêt à rester à l’hombre de Warburg…