Publication d’un essai préclinique entrepris à l’initiative de la Fondation Guérir du Cancer
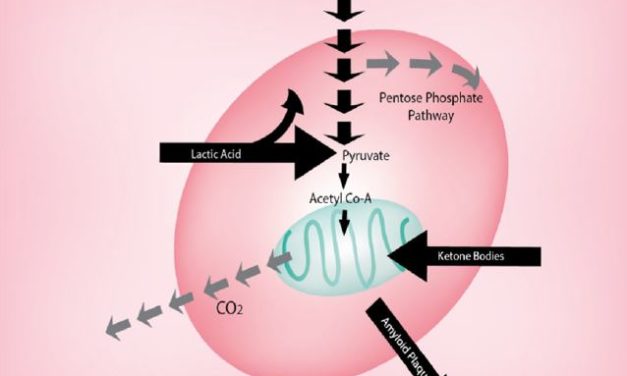
Fondation Guérir du Cancer – Février 2024 Publication d’un essai préclinique entrepris à l’initiative de la Fondation Guérir du Cancer Grâce à la générosité de nombre d’entre vous, la Fondation Guérir du Cancer,...
En savoir plus





Commentaires récents